
Desktop Applications
With the odd exception, the desktop applications produced by the ICS group are available across a range of platforms and are designed to be both easy-to-use and easy-to-deploy. We provide simple installers (via the excellent install4j suite) that bundle both the application and any of its dependencies in a single package.
You can find summary information below, as well as links to each application’s home page where they are described in more detail.
Humbug
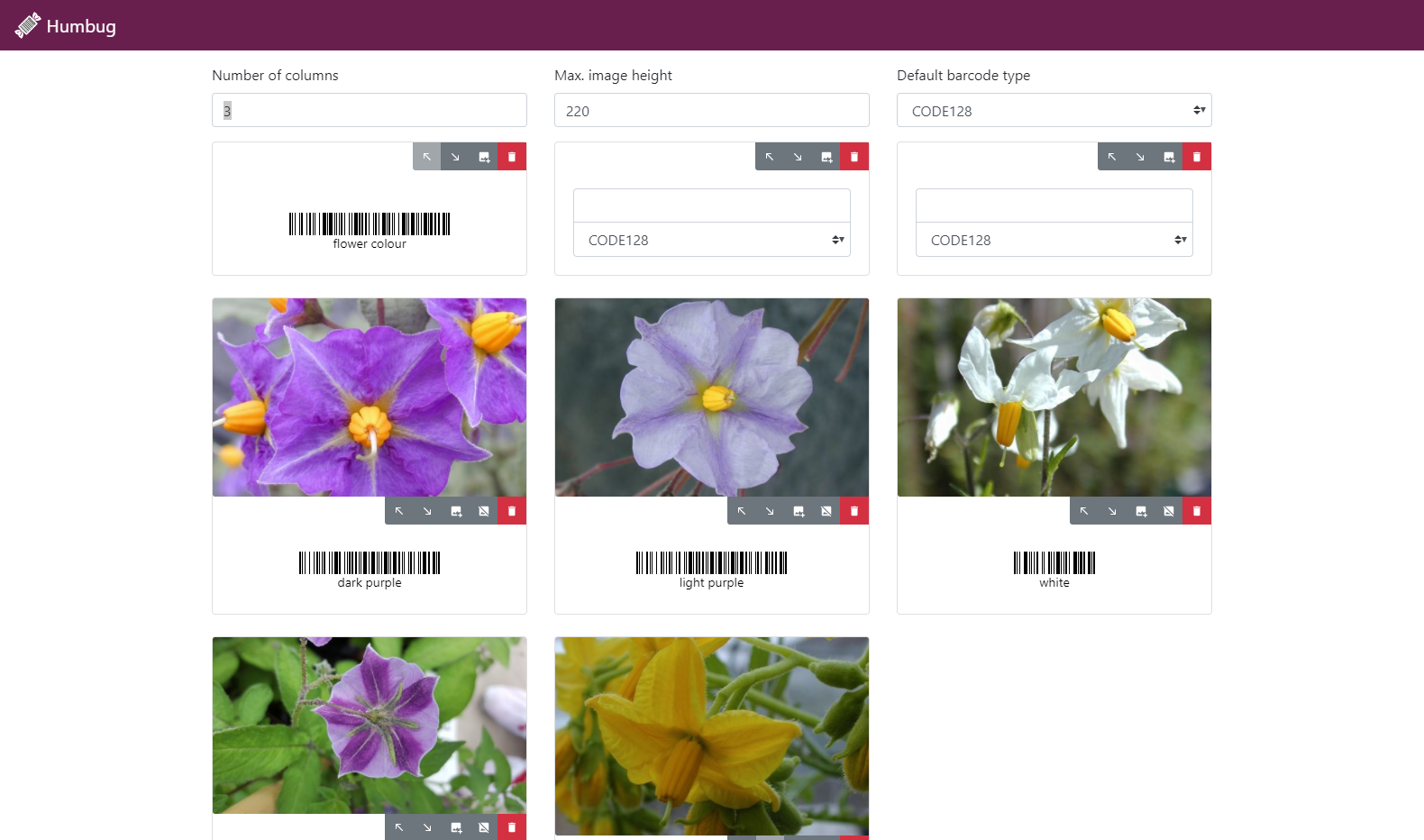 Humbug is a tool to generate sheets of user-defined barcodes to aid in the recording of data in the field or glasshouse. Humbug users define these categories using a simple to use and intuitive desktop application then print out sheets containing a barcode for each data category required. These barcodes can then be scanned using Germinate Scan and saved to text file.
Humbug is a tool to generate sheets of user-defined barcodes to aid in the recording of data in the field or glasshouse. Humbug users define these categories using a simple to use and intuitive desktop application then print out sheets containing a barcode for each data category required. These barcodes can then be scanned using Germinate Scan and saved to text file.
|
More information: |
https://ics.hutton.ac.uk/get-humbug |
|
Platforms supported: |
|
|
Primary developers: |
Sebastian Raubach and Paul Shaw |
Germinate Daim
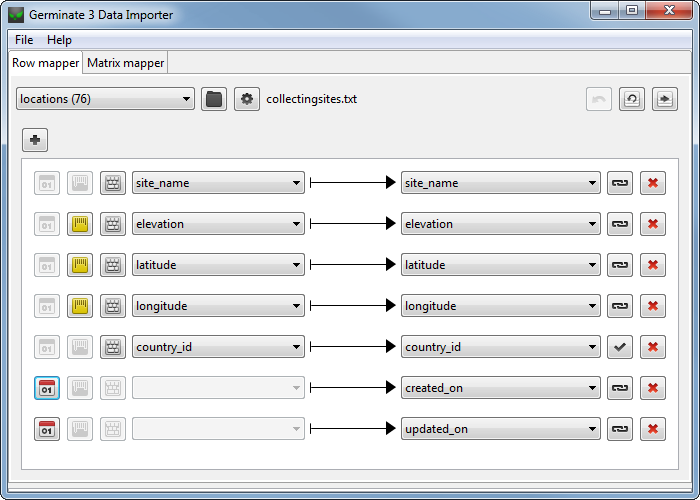 Germinate Daim is a desktop application that simplifies the process of importing data into the Germinate database by providing a straight-forward and easy-to-use interface.
Germinate Daim is a desktop application that simplifies the process of importing data into the Germinate database by providing a straight-forward and easy-to-use interface.
|
More information: |
https://ics.hutton.ac.uk/germinate-daim |
|
Platforms supported: |
|
|
Primary developers: |
Sebastian Raubach and Paul Shaw |
Helium
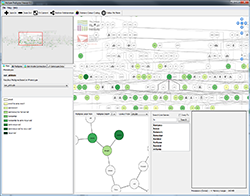 Plant breeders utilise a large number of varied and diverse data types in order to choose suitable plant lines for subsequent breeding generations to ensure that characteristics of agricultural importance are maintained and improved in commercially grown varieties. With recent technological advances, the ability to generate large experimental data sets is becoming both increasingly cheaper and easier to perform. This however leads to problems in data handling and visualization for end users. The ability to merge these data together and provide appropriate visualizations using a pedigree structure based framework and supporting database back-end will allow breeders and researchers to make better decisions choosing potential crosses to carry out in order to aid development of new cultivars.
Plant breeders utilise a large number of varied and diverse data types in order to choose suitable plant lines for subsequent breeding generations to ensure that characteristics of agricultural importance are maintained and improved in commercially grown varieties. With recent technological advances, the ability to generate large experimental data sets is becoming both increasingly cheaper and easier to perform. This however leads to problems in data handling and visualization for end users. The ability to merge these data together and provide appropriate visualizations using a pedigree structure based framework and supporting database back-end will allow breeders and researchers to make better decisions choosing potential crosses to carry out in order to aid development of new cultivars.
|
More information: |
https://ics.hutton.ac.uk/helium |
|
Platforms supported: |
|
|
Primary developers: |
Paul Shaw |
Tablet
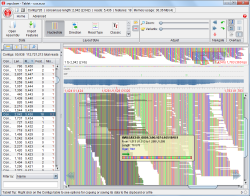 Tablet is a high performance graphical viewer for second-generation sequence assemblies and alignments. It allows users to easily visualize their read data against reference data, including scaled-to-fit and coverage overviews and an extremely flexible main display allowing for visualization of the data at many different resolutions. Tablet supports all major assembly formats including the now de-facto standard SAM/BAM format, as well as the GFF3 format for importing of supplementary annotation data. The application is recognised externally as an excellent tool for the visualization of sequence assemblies; several groups now bundle Tablet with their software suite as the assembly viewer of choice.
Tablet is a high performance graphical viewer for second-generation sequence assemblies and alignments. It allows users to easily visualize their read data against reference data, including scaled-to-fit and coverage overviews and an extremely flexible main display allowing for visualization of the data at many different resolutions. Tablet supports all major assembly formats including the now de-facto standard SAM/BAM format, as well as the GFF3 format for importing of supplementary annotation data. The application is recognised externally as an excellent tool for the visualization of sequence assemblies; several groups now bundle Tablet with their software suite as the assembly viewer of choice.
|
More information: |
https://ics.hutton.ac.uk/tablet |
|
Platforms supported: |
|
|
Primary developers: |
Iain Milne, Gordon Stephen, Micha Bayer, and Linda Milne |
Flapjack
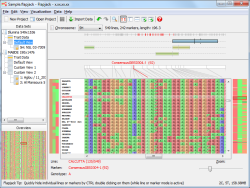 Flapjack provides interactive visualizations of high-throughput genotype data, allowing for rapid navigation and comparisons between lines, markers and chromosomes. It includes graphical views of alleles coloured by state, frequency or similarity to a given line, along with visualizations of phenotypic and QTL data, and marked-based plots. It supports a range of interactions, including graphically moving lines or markers, insertions or deletions of data, and sorting or clustering of lines by either genotype similarity to other lines, or by trait scores. Any map based information such as QTL positions can be visually aligned against graphical genotypes to identify associated haplotypes.
Flapjack provides interactive visualizations of high-throughput genotype data, allowing for rapid navigation and comparisons between lines, markers and chromosomes. It includes graphical views of alleles coloured by state, frequency or similarity to a given line, along with visualizations of phenotypic and QTL data, and marked-based plots. It supports a range of interactions, including graphically moving lines or markers, insertions or deletions of data, and sorting or clustering of lines by either genotype similarity to other lines, or by trait scores. Any map based information such as QTL positions can be visually aligned against graphical genotypes to identify associated haplotypes.
|
More information: |
https://ics.hutton.ac.uk/flapjack |
|
Platforms supported: |
|
|
Primary developers: |
Iain Milne, Gordon Stephen, and Paul Shaw |
CurlyWhirly
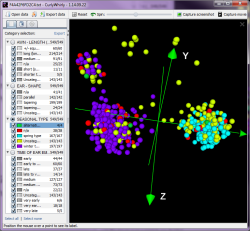 CurlyWhirly is a graphical, standalone software tool for examining graph data in 3D, for example the output from principal components analyses. It is implemented in Java 3D and Swing and allows selective highlighting of categories so users can easily display subsets of their data and, for example, detect outliers.
CurlyWhirly is a graphical, standalone software tool for examining graph data in 3D, for example the output from principal components analyses. It is implemented in Java 3D and Swing and allows selective highlighting of categories so users can easily display subsets of their data and, for example, detect outliers.
|
More information: |
https://ics.hutton.ac.uk/curlywhirly |
|
Platforms supported: |
|
|
Primary developers: |
Gordon Stephen, Iain Milne, and Micha Bayer |
Strudel
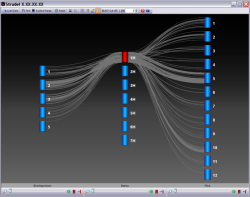 Strudel is an advanced graphical tool for visualizing genetic and physical maps of genomes for comparative purposes. The application aims to let the user examine their data at a variety of different levels of resolution, from entire maps to individual markers, and explore syntenic relationships between genomes. All browsing and interaction with Strudel happens in real-time – there is no need to wait while the maps are generated as it often the case with server-side map applications.
Strudel is an advanced graphical tool for visualizing genetic and physical maps of genomes for comparative purposes. The application aims to let the user examine their data at a variety of different levels of resolution, from entire maps to individual markers, and explore syntenic relationships between genomes. All browsing and interaction with Strudel happens in real-time – there is no need to wait while the maps are generated as it often the case with server-side map applications.
|
More information: |
https://ics.hutton.ac.uk/strudel |
|
Platforms supported: |
|
|
Primary developers: |
Micha Bayer and Iain Milne |
TOPALi v2
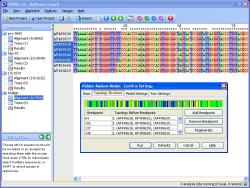 Developed in collaboration with Biomathematics & Statistics Scotland (BioSS), TOPALi is a graphical application that simplifies and automates the use of several methods for the evolutionary analysis of multiple sequence alignments. Analysis jobs are submitted from a local client to run either as web services on the high-performance compute cluster at the James Hutton Institute, or locally on the user’s desktop. Methods available include model selection and phylogenetic tree estimation using the Bayesian inference and maximum likelihood (ML) approaches, in addition to recombination detection methods.
Developed in collaboration with Biomathematics & Statistics Scotland (BioSS), TOPALi is a graphical application that simplifies and automates the use of several methods for the evolutionary analysis of multiple sequence alignments. Analysis jobs are submitted from a local client to run either as web services on the high-performance compute cluster at the James Hutton Institute, or locally on the user’s desktop. Methods available include model selection and phylogenetic tree estimation using the Bayesian inference and maximum likelihood (ML) approaches, in addition to recombination detection methods.
|
More information: |
http://www.topali.org |
|
Platforms supported: |
|
|
Primary developers: |
Iain Milne and Frank Wright (BioSS) |
TOPALi v1
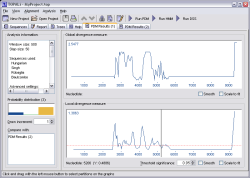
|
More information: |
http://www.topali.org/topali-v1 |
|
Platforms supported: |
|
|
Primary developers: |
Iain Milne and Frank Wright (BioSS) |
TetraploidMap
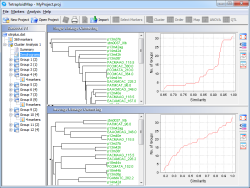 Developed in collaboration with Biomathematics & Statistics Scotland (BioSS), TetraploidMap is a graphical user interface for calculating linkage maps for autotetraploid populations. It is suitable for handling markers scored on two parents and the full-sib offspring of a cross between them. TetraploidMap handles both codominant and dominant molecular markers, in all possible configurations, and takes into account the presence of null alleles in the analysis. It now includes a routine for QTL mapping.
Developed in collaboration with Biomathematics & Statistics Scotland (BioSS), TetraploidMap is a graphical user interface for calculating linkage maps for autotetraploid populations. It is suitable for handling markers scored on two parents and the full-sib offspring of a cross between them. TetraploidMap handles both codominant and dominant molecular markers, in all possible configurations, and takes into account the presence of null alleles in the analysis. It now includes a routine for QTL mapping.
|
More information: |
http://www.bioss.ac.uk/knowledge/tetraploidmap |
|
Platforms supported: |
|
|
Primary developers: |
Iain Milne and Christine Hackett (BioSS) |
LandSFACTS
The LandSFACTS model facilitates the creation of spatial land use change scenarios under climate change. The latest developments include shaping the land use changes to enhance multiple ecosystem services, while considering land capability and farm types across the landscape. The model provides a framework to explore fully spatialised land use scenarios to test hypotheses supporting land use policies explorations, and to then be able to assess their potential impacts on Ecosystem Services (e.g. carbon balance, nutrient exports, connectivity).
|
More information: |
https://ics.hutton.ac.uk/landsfacts |
|
Platforms supported: |
|
|
Primary developers: |
Marie Castellazzi |